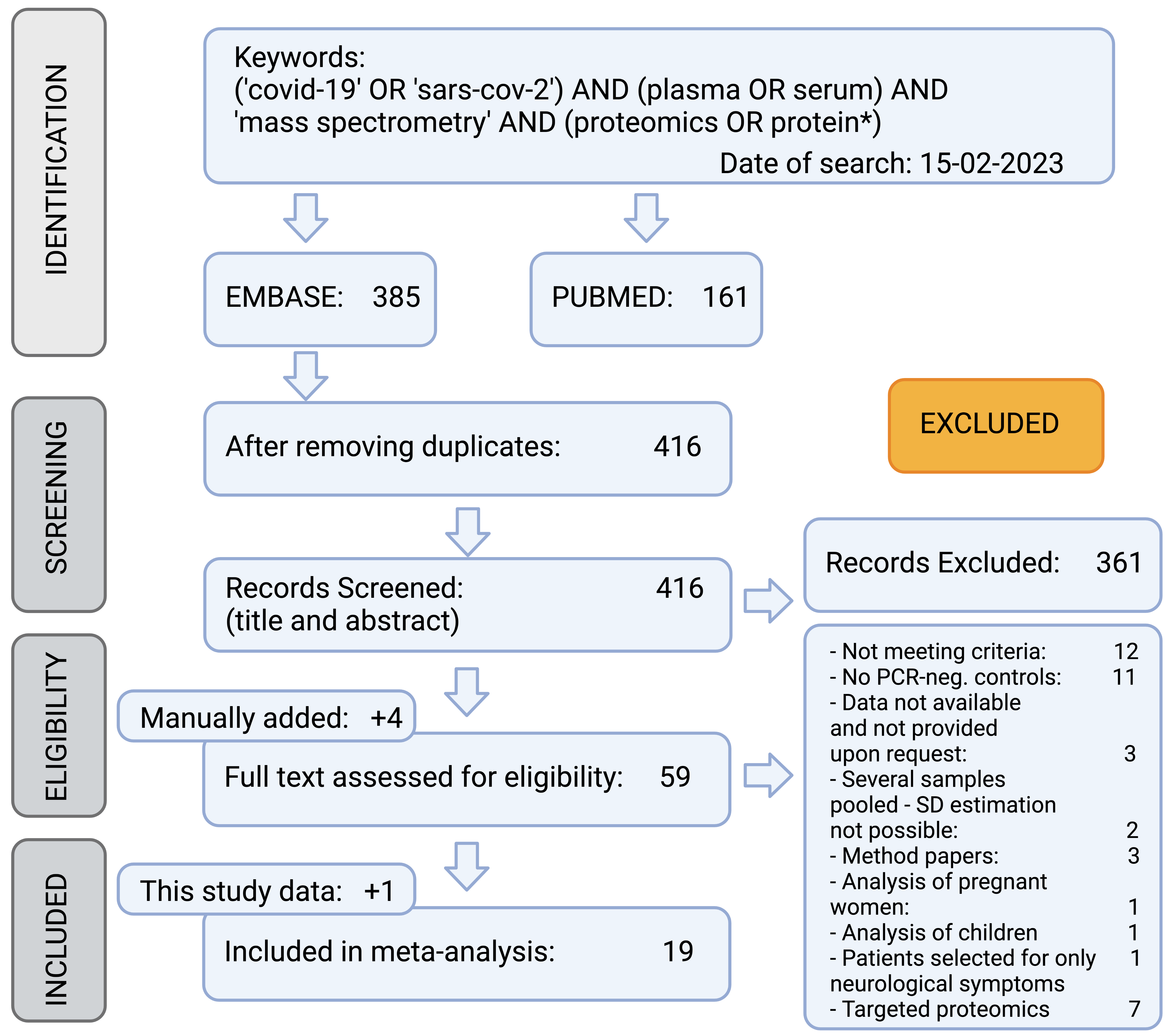
SROC curve for selected protein
The summary receiver operating characteristics (SROC) curve summarises the sensitivity and specificity estimates obtained in different studies.
It is calculated according to the bivariate model of
Reitsma J et al. (2005. Journal of Clinical Epidemiology).
using
a generalised linear mixed model.
The Meta-Analysis (MA) Estimate point presents the mean true positive rate (TPR, sensitivity) and mean false positive rate (FPR, 1-Specificity).
The positive test was determined based on estimating an optimal cut-off point per protein in each study, to determine the optimal trade-off between sensitivity and specificity.
Corresponding 95% confidence intervals (CI) of TPR and FPR and 95% CI ellipsoid area in which the expected true estimate lies are also plotted.
Finally, Diagnostic odds ratios (DOR) based on Reitsma's bivariate model (with 95% CI), and a summary area under the curve (AUC) estimate is provided in the plot.
As a rule of thumb, a DOR of value 10 and above and AUC of 90% and above indicate an excellent biomarker. DOR estimates with 95% confidence intervals that do not contain 1 are statistically significant.
If the 95% CI and ellipsoid area are not extending beyond the Chance line, the estimates are statistically-significant and such a protein can be useful as a biomarker of COVID-19.
We further estimated the preference of the study-specific ROC curves for sensitivity and specificy, following the approach by
Doebbler & Holling (2015).
The plot does not depict whether the protein is higher or lower in COVID-19.
If you are interested in that, check under the Protein SMD tab.
Forest plots for selected protein
The forest plots present the DOR, sensitivity and specificity estimates for selected protein.
For transparency, all proteins that are identified in the analysis are plotted, regardless of whether sigificant or not.
Priority should be given to the estimates of SROC curves, DOR, sensitivity and specificity provided by the bivariate Reitsma model.
Please note that estimates for proteins identified in fewer studies are less reliable.
Estimates based on only two studies should be considered unreliable.
Sensitivity
Sensitivity is the proportion of true positive tests among those who had the disease - COVID-19 in this instance.
The graph below contains all the sensitivity estimates per study, with 95% CI. The summary estimate is based on the bivariate Reitsma model.
High sensitivity (>0.8) implies that the protein can be a useful diagnostic biomarker of COVID-19 patients, meaning that most COVID-19 patients should have altered levels of this protein.
Specificity
Specificity is the proportion of true negative tests among those who did not have the disease - PCR-negative individuals in this instance.
The graph below contains all the specificity estimates per study, with 95% CI. The summary estimate is based on the bivariate Reitsma model.
High specificity (>0.8) implies that the protein can be a useful biomarker for excluding a diagnosis of COVID-19.
Diagnostic odds ratios
The Diagnostic odds ratio (DOR) represent the ratio between the odds od testing positive in individuals with COVID-19 and the odds of testing negative in individuals who were PCR-negative in the corresponding studies.
The summary DOR in this plot was estimated with a univariate method
(Glas et al. Journal of Clinical Epidemiology).
Proteins with 95% CI of log(DOR) that do not overlap 0 are statistically significant.
As a rule of thumb, a DOR of value 10 and above (corresponding to a log value of 2.23 in the plot) indicates a very good biomarker.